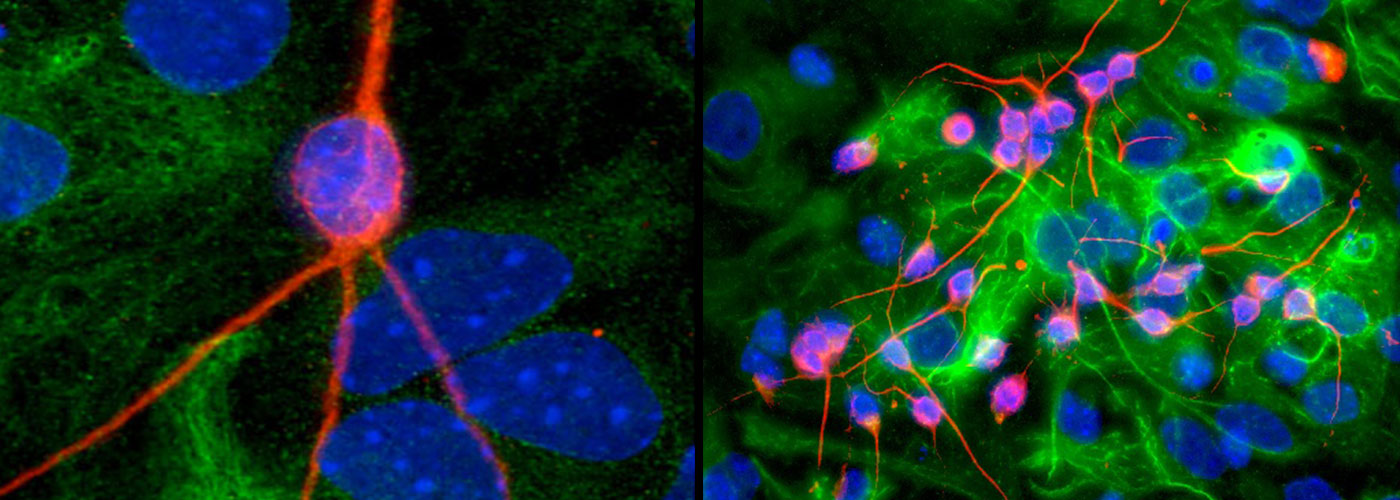
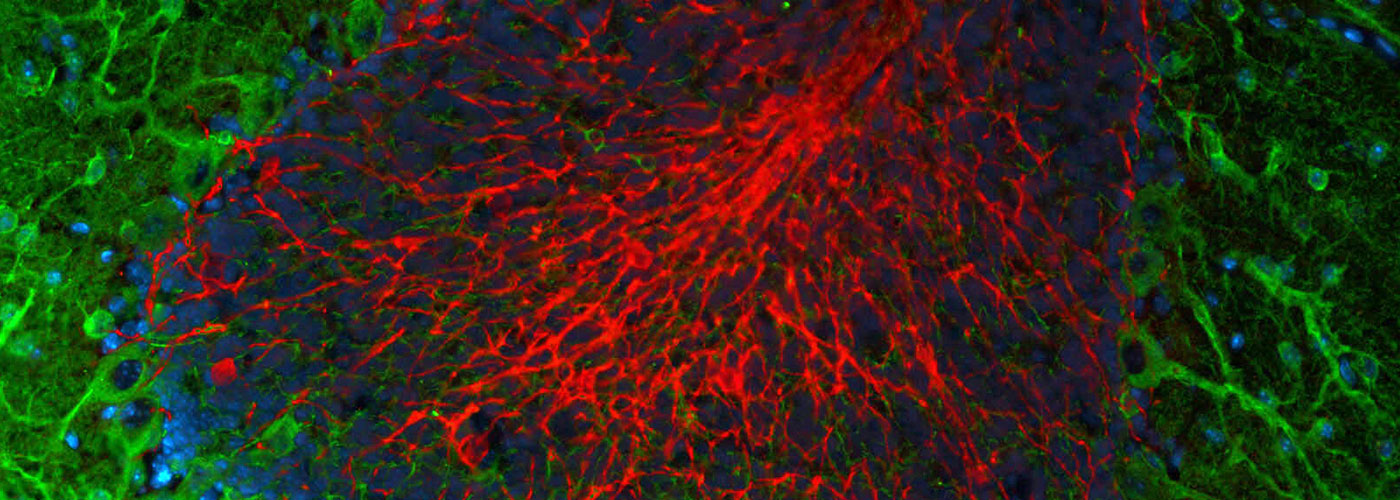
 Weekly reads: MYC phosphorylation, reverse protein aging, mitochondria don’t like space, that peptide committee
Weekly reads: MYC phosphorylation, reverse protein aging, mitochondria don’t like space, that peptide committeeWhy am I interested in MYC phosphorylation? What is MYC? Long-time readers of The Niche probably remember that the MYC family is one of my primary interests. These proteins are essential for normal development (and stem cell function) as well as contributing to the formation of many tumors when overexpressed…

Potentially highly-conflicted PCAC meets this week on pop peptides, but no waivers
So-called wellness peptides are even more in the spotlight this week as the pharmacy compounding advisory committee or PCAC launches …
Weekly reads: T cell stem cells, thymus as anti-aging organ, octopus ribosomes, longevity ethics
When immunology researchers talk about “T cell stem cells” (or stem T cells or T memory stem cells), are they …
Human embryo models (SCBEM) are powerful but probably won’t get a Nobel; lessons about rewards in science
There has been some discussion that the next-generation research on human stem cell-based embryo models (SCBEM) could someday warrant a …
Weekly reads: Colossal Bio & Trump admin partnership, FDA commish finalists, appetite-regulating neurons
Readers of The Niche will recall that I’ve been skeptical of the efforts of the de-extinction firm Colossal Bio. For …
Help support our pediatric brain tumor research

Visit Our Facebook Page

Visit our Stem Cell Channel on YouTube

Dr. Knoepfler’s TED talk
